Suggestions¶
In this page you will find some basic suggestions about the Geomosaic execution that may help you. Over time, we plan to update this page based on our experience and the open issues.
Create your workflow in two major steps¶
The issue I will explain here arises only when creating a workflow that includes modules for MAG annotation. There are two possible solutions to address this problem:
use
geomosaic unitfor each module of your interestdivide your
geomosaic workflowinto two major steps
Before going into the details, let me explain this issue. I suggest you read it all and do not skip straightly to the solutions section.
Issue: No such file or directory: /path/to/MAGs.tsv¶
When you try to create a workflow from scratch to modules related to the MAGs annotation (all the modules after the mags_retrieval, for example mags_orf_prediction), it happens that you will get an error related to a file called MAGs.tsv. Basically, all modules related to the MAGs annotation require a file called MAGs.tsv, which however is created by the mags_retrieval.
Indeed, before running the workflow, Snakemake tries to check if it has all the expected files to start the computation. However, in the above case, Snakemake is expecting to have a MAGs.tsv file, which cannot be found because it is planned to be created within the mags_retrieval module (rule named checkpoint run_mags).
This is a technical issue that comes from how SnakeMake works, which is not fully compatible with how I need it to work on (possible) multiple MAGs (you can read more about this here: https://github.com/snakemake/snakemake/issues/2523). The solution that I had to implement in Geomosaic is not completely good in this scenario. However, as I said, there are two possible solutions that the user can adapt.
Solution #1: use geomosaic unit for each module of your interest¶
The first solution concerns the use of geomosaic unit instead of the geomosaic workflow, thus executing one module at a time. In this way, Snakemake will only checks the requirements for a single module.
From personal experience, I like using the geomosaic unit as it allows me to have more control over each module, check the status of each job, check if it had some errors on some samples, and ignore them in the next steps etc…
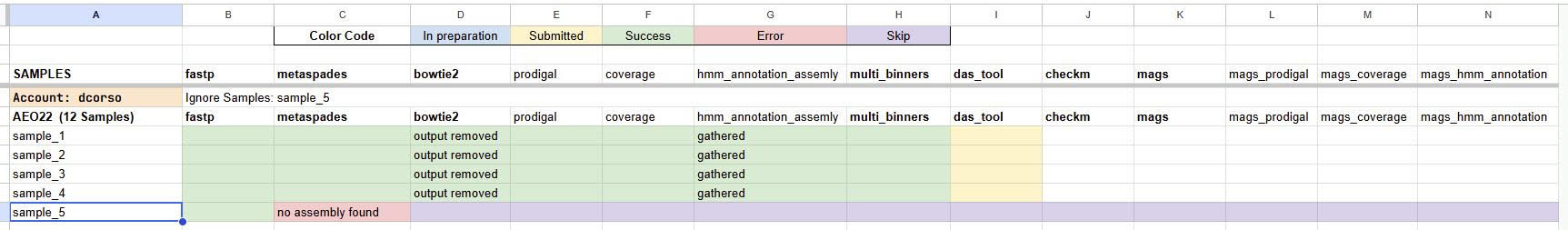
For example, this image shows how I’m controlling all the modules while executing the unit command since I’m executing one module at a time, and then checking the logs to see if some samples failed. In this case, I can ignore specific samples for future modules in the geomosaic prerun. This may be a solution on controlling the modules that you are executing. Keep in mind to do a new geomosaic prerun after each unit, specifying the flag --unit and also putting a specific folder for the logs. For example, if I’m executing the package das_tool (which belongs to the binning_derep module), in the prerun command I put the following flag -f slurm_logs/das_tool
Solution #2: divide your geomosaic workflow into two major steps¶
The second solution is to divide the workflow into two major steps (or two sub-workflows).
In the first step, you should execute a
geomosaic workflowthat has all the modules that you want up to themags_retrievalincluded. No more than this.When all these modules are executed, you can create another
geomosaic workflowstarting from themags_retrieval(using the corresponding flag--module_start <modulename>). With this step you can execute all the modules that you want on the Stream Binning-level, such asmags_orf_prediction,mags_tax_annotationand so on.
Why these two steps?
The first workflow is useful to create the MAGs.tsv, so snakemake won’t check for its existence for the module like mags_orf_prediction (since are not there). Once this workflow is successfully executed, and so the file MAGs.tsv is created, the second workflow will try to check the existence of this file, and since it will find it you won’t get any error like the one above.
Create a specific log folder for each tool¶
When executing the geomosaic prerun, we suggest putting a specific folder for the tool that we are running or for our workflow. For instance, if we are running the assembly using metaspades with the geomosaic unit command, in the prerun you can use -f slurm_logs/metaspades to put all the corresponding logs in that folder.
Delete corresponding output folder before re-executing a tool¶
If you would like to execute a tool again in a specific module, we suggest deleting the previous output folder from sample directories or renaming it. Indeed, snakemake can check if the output does exist, and in this case, it avoids rerunning the computation.
Execute commands with geomosaic conda environment activated¶
The geomosaic conda environment must be activate before each command, even when submitting jobs through SLURM or executing the workflow using GNU Parallel.
Execute geomosaic commands in the same folder that contain gmsetup.yaml¶
In the Walkthrough Tutorial you can see that each command was executed in the same folder where the gmsetup.yaml was. We proposed this organization to work inside the root folder of the geomosaic computation of your dataset.
[For Expert Users] Execution types of the workflow¶
To execute the created workflow, you can use the prerun command to create scripts for SLURM or GNU Parallel. When available we always suggest to use --exec_type slurm with the prerun.
However, since the workflow is a Snakefile, if you are familiar with Snakemake you can also use it to run the workflow. You will need to specify the following flags
--use-conda--conda-prefix <path to the geomosaic conda envs folder>
You can always refer to our geomsoaic templates
Moreover, Snakemake has its own tutorial to execute the workflow using SLURM.