Integration example 1: Simple package¶
This tutorial will guide you through an integration of a simple package, that doesn’t need an external database and it is not referred to MAGs module.
What we need for this integration¶
Understand the Stream-level: in this case
Read-basedModule name:
reads_qcPackage name:
fastqc_readscountKnow which are the conda dependencies for this packages, or the conda package name.
Know the code to actual integrate and execute the package.
Step 1: Clone/fork the repository and install Geomosaic¶
Since the final strategy is to make a pull request to the main repository, we suggest to fork our repo and then clone it (in the SSH way)
git@github.com:<YOURNAME>/Geomosaic.git
Install the Geomosaic conda environment. You can follow the Installation Guide.
Remember to replace <YOURNAME> with your GitHub user account.
Once you have cloned the repository, open the directory created with the clone and also create another branch specifying the name of the package that you are going to integrate
git checkout -b fastqc
Step 2: Create the module folder (if does not exists)¶
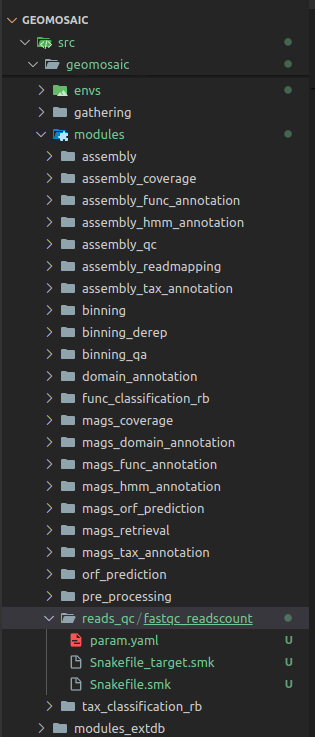
In this case we are going to integrate a package that belongs to a module related to the quality checks of the reads after the pre_processing step, so we create a module folder called reads_qc inside the modules folder (Figure below in Step 4).
Important
This step is necessary only if the module folder does not exists.
Warning
Do not use any special characters or insert spaces in the name.
Highlight
Just rely on underscore and all lower-case characters
Step 3: Create the package folder¶
In this step we create the package folder inside the module of interest. In this case, our package folder will be fastqc_readscount (Figure below in Step 4).
Warning
Do not use any special characters or insert spaces in the name.
Highlight
Just rely on underscore and all lower-case characters
Step 4: Create package’s snakefiles¶
Create three code files inside the package folder, with the following filename:
Snakefile.smkSnakefile_target.smkparam.yaml
For now you can leave them empty.
Important
The names for this file are standard and are the same for each package. Do not change the filenames.
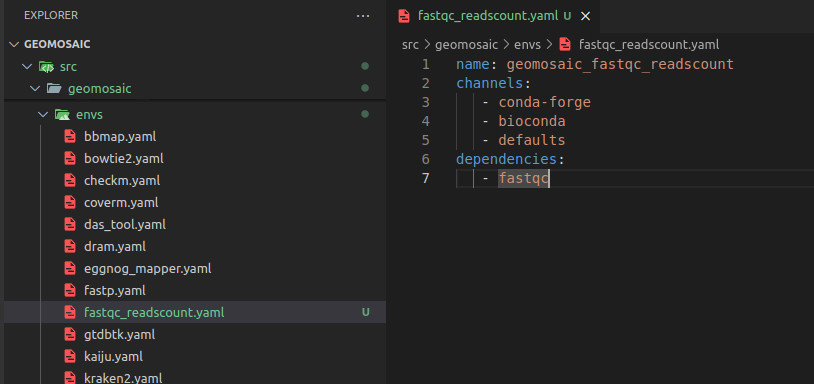
Step 5: create the corresponding conda env file¶
We need to create the corresponding conda env file describing the necessary dependencies for our package. For this purpose, in the envs folder we create a file with the same name of the package (with the yaml extension). As content of the file we are going to write the necessary dependencies. In this case we are going to specify only fastqc from the bioconda channel as the reads count will be computed through a bash commands and thus we don’t need any specfici dependencies. The name of the conda environment is the name of the package with geomosaic_ as prefix.
Now we can write our code inside the Snakefile
Step 6: Link the module/package to the Geomosaic core¶
So in this section we need to link our new module and/or our new package to the already existing core of Geomosaic, which is represented by the file called gmpackages.json
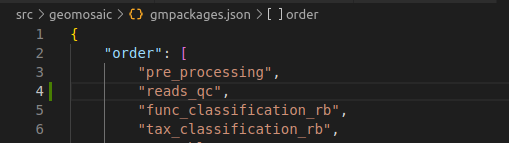
Step 6.1: order section¶
Since reads_qc is a module that we thought to be after the processing of the reads, we put it after pre_processing but before the assembly.
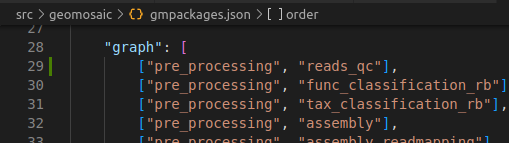
Step 6.2: graph section¶
Important
Before going further in this section, you should understand what really means a dependency in Geomosaic in this Modules Dependencies description.
The package that we are going to integrate in this module, depends on the output reads obtained from the pre_processing modules, so we put in graph the following line:
["pre_processing", "reads_qc"],
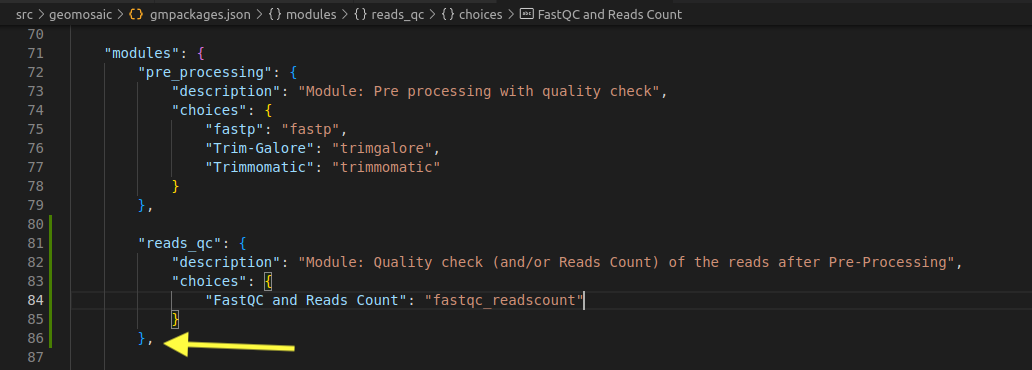
Step 6.3: modules section¶
In the correspongin modules section, we need to add the name of the module, which then must contain the following two keys
description- which contains a brief description of the modulechoices- which is a dictionary containing all the packages belonging to that module.
In particolar, the key (the blu string in the image) is the String that will come out in the terminal as a choice, during the workflow decision, while the value (in orange) is the actual name of the package, the one that we used also to create the folder created in step 3.Important
Package name on the value must match with the folder created in the step 3
Important
Remember the last comma after the last parenthesis.
Step 6.4: additional_input section¶
If the package does require any additional input, you can integrate this input in the corresponding section of additional_input. In this case we don’t need to put any additional argument.
Highlight
Additional arguments are parameters that are widely known in the metagenomic workflow and that should be chosen by the user, as for example Completeness and Contamination.
In this section we have inserted also the possibility to specificy a folder that contains HMM models (for assembly_hmm_annotation and mags_hmm_annotation), as well as the name of the output folder these two modules in order to have different output name folder for different sets of HMMs.
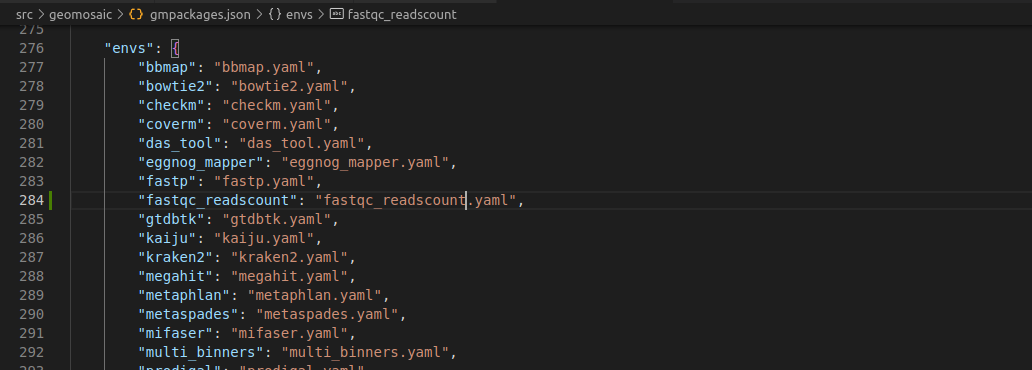
Step 6.5: envs section¶
This section is very simple, we only need to add the conda env file for our package. This filename must have the same package name. In this case fastqc_readscount.
Step 6.6: external_db section¶
This section is useful to organize external databases for the package that we are going to integrate. In this example, we won’t need any external database. Look to this example to understand how this section works.
However, let’s do a brief introduction to this section:
each package that requires an extdb has a key which contains two other keys:
inpfolder: its value should be the name of the packageoutfolder: must be the name of the folder in which the external databases is going to be downloaded. The pattern is: the name of the package followed by the_extdbsuffix. However, different package name maybe relay on the same external database as it is for therecognizerpackage andmags_recognizer, therefore in such case we specify the sameoutfolder.
For this package we don’t need to specify anything for external database
Step 6.7: gathering section¶
This section is not mandatory. However it is useful if we want to compose some master tables or plots from the results of our package from all the samples. In this case, we are not interest in this section. However we could create a script in which we create a table of all the reads count for each sample.
At the time of writing, this one is the last section.
Step 7: Write the actual code.¶
For this package the code is very easy. Since it uses only the processed reads, we can use the template of the assembly. We copy paste the code inside the Snakefile.smk of metaspades and then modify it.
Step 7.1 Snakefile: input/output section¶
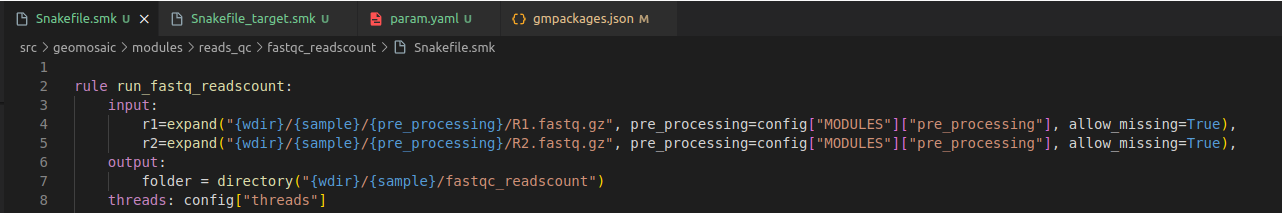
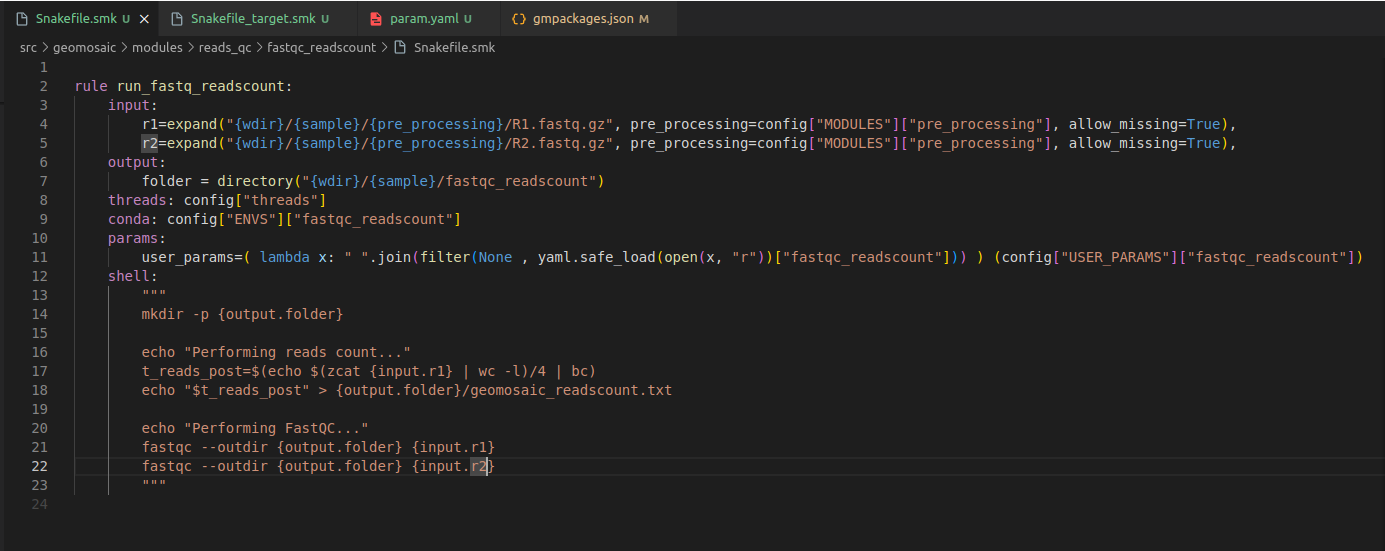
We need to change the rule definition with the package name, composed also of the prefix run_.
Our input section is fine, as we need only the reads from the
pre_processingmodule.In output section usually we put the folder output that must be the same of the package name. However if you know that your package is going to provide in output a specific file, you can even increase the detail of this section by inserting also that file.
Step 7.2 Snakefile: threads section¶
The threads section is fine like this. If we know that is not possible to execute our package through parallelization we can put in this section 1, otherwise we can leave it as it is.
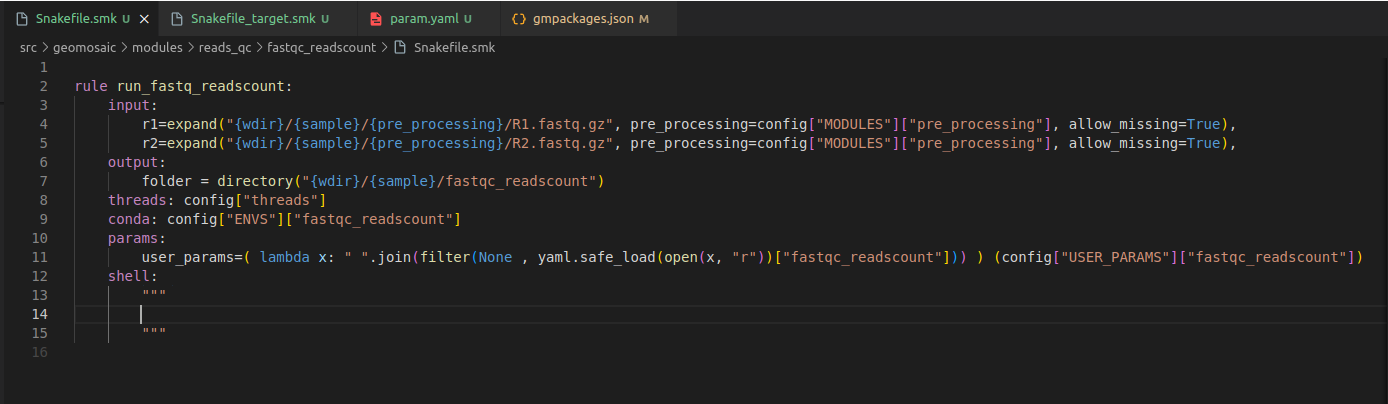
Step 7.3 Snakefile: conda section¶
In this section, we only need to put our package name.
Step 7.4 Snakefile: params section¶
In each package we put a params variable called user_params, which is going to read the param.yaml file that we have created in the Step 4. The code to read user parameters, is almost always the same (so you don’t need to modify it):
user_params=( lambda x: " ".join(filter(None , yaml.safe_load(open(x, "r"))["fastqc_readscount"])) ) (config["USER_PARAMS"]["fastqc_readscount"])
Just replace fastqc_readscount with your package name.
Step 7.5 Snakefile: shell section¶
This is the section in which we are going to put the actual code to execute our programs.
So for this package we want to integrate the use of fastqc to check the quality of the reads, and then perform a reads count on the processed reads.
Step 8: Snakefile Target¶
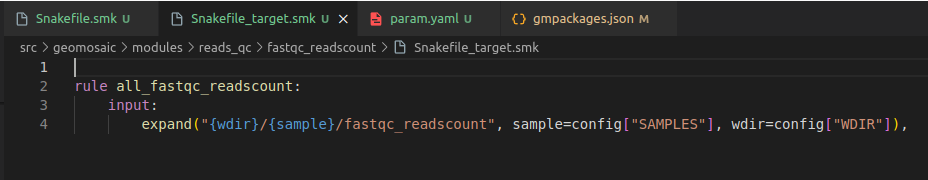
In our file Snakefile_target.smk we only need to write few rows. First, the name of the rule must be the same name of the package name with the all_ prefix. And then we need to change the rows in the input section, and we need to specify the same folder output as in this case was our only output that we specified in the Snakefile.smk.
Step 9: Param.yaml file¶
The param.yaml is a file in which the user, before the execution of the workflow, can insert all the optional parameters belonging to the package as bullet points. In this case, we only need to open this file and add the following line at the top:
fastqc_readscount:
-
Test the integration¶
Now we should test the integrated package. Activate the conda environment of geomosaic. Updated geomosaic by doing
pip install .
Once we have tested, we can commit the changes and create the pull request.